
In the present work a simple and fast microanalytical colorimetric method using diphenyl amine chromogenic reagent was worked out and validated for the quantitative determination of alpha-1-acid glycoprotein (AGP, orosomucoid) in human serum. Sample preparation based on non-miscible phase solvent extraction and precipitation in the form of metal ion (Zn) complex were applied to the mild and specific isolation of AGP. In order to quantitate AGP on the base of its carbohydrate content chromogenic reagents, phenol-sulphuric acid, orcine-sulphuric acid and diphenyl amine used generally in the colorimetric measurement of mono- and oligosaccharides were compared. Experimental conditions of the diphenyl amine color reaction were studied in details and optimized. Investigating the chromogenic properties of AGP monosaccharide constituents with diphenyl amine it could be stated that only galactose and mannose resulted in measurable but different specific absorption at 630 nm. Reversed phase HPLC methods were applied to determine the level and molecular distribution of monosaccharides in the oligosaccharide structure of AGP. Carbohydrate analysis of individual AGP samples isolated from the sera of healthy persons and cancer (ovary and uterus) patients showed that, in general, galactose and mannose constitute altogether the 15.5% of AGP by weight, with a molar ratio of 1.35. Based on this observation the applicability of a standard (stock) solution containing 57.5% of galactose and 42.5% of mannose was proved to be identical with AGP as reference compound for the calibration of the colorimetric method. On the other hand, the validity of the colorimetric method was supported by the parallel determination of AGP concentrations in individual serum samples using the reference ion exchange chromatographic procedure. Finally, determination of serum AGP level in a population of 75 healthy persons resulted in a mean of 59.9±16.2 mg/dl. Statistical analysis of data showed 89% specificity for normal values at a cut-off level of 80 mg/dl suggested.

A rákbetegség morbiditási és mortalitási adataiból, valamint az erre vonatkozó klinikai-laboratóriumi vizsgálatok számának és minőségének alakulásából általában megállapítható, hogy a rosszindulatú daganatos megbetegedések száma világszerte növekszik és ezzel összefüggően fokozódik az igény az újabb diagnosztikai és monitorizálási eljárások fejlesztésére és alkalmazására (1-7). Ugyancsak megfigyelhető, hogy a marker kutatások egyre nagyobb mértékben alkalmazzák a korszerű, nagy felbontó képességű és érzékenységű mikroanalitikai és elválasztástechnikai eljárásokat (HPLC, kapilláris elektroforézis, tömeg~spektrometria) (8-11). A számos, jelenleg ismert és alkalmazott daganat marker közül a szénhidrát tartalmú komplex fehérjék, az ún. szialoglikoproteinek iránt diagnosztikai jelentőségük, valamint oligoszacharid szerkezetük sajátosságai miatt az érdeklődés egyre növekszik (16-19). Ezek a biopolimerek szerkezetileg kötött, élettani funkcióik szempontjából nélkülözhetetlen szénhidrátokat, elsősorban neutrális monoszacharidokat, hexózaminokat és sziálsavat tartalmaznak. A szialoglikoproteinek molekula szerkezetének vizsgálata kimutatta, hogy szénhidrát tartalmukat a peptid lánc aszparagin, ill. szerin treonin aminosav szekvenciáihoz N-, ill. O-glikozidos kötésekkel kapcsolódó, ún. antennáris szerkezetű oligoszacharid láncok (glikán egységek) alkotják (8,10,19,20). Az oligoszacharid láncok monoszacharid összetevőinek arányai és sorrendisége, a láncok hossza, elágazásai és terminális sziálsav tartalma a lehetséges molekula variánsok nagy változatosságát eredményezi. A szialoglikoproteinek oligoszacharid összetételének és lettani funkcióinak összefüggései alapján feltételezik, hogy antennáris szerkezetükben olyan biológiai kódrendszer (sugar language) ismerhető fel, amely a fehérjék glikozilálásának poszttranszlációs folyamatai során endogén és/vagy exogén tényezők hatására kialakult molekuláris információkat tartalmaz. A legtöbb biológiailag aktív, strukturális és funkcionális szempontból alapvetően fontos vegyületről (pl. vércsoport (Lewis) antigének, interferonok, erythropoietin, membránfehérjék, stb) és szinte minden eddig ismert és a gyakorlatban általánosan alkalmazott konvencionális tumor markerről (CEA, AFP, TPA, hCG, NSE, CA 125, CA 15-3, CA 72-4, CA 19-9, akut fázis proteinek, stb) kimutatták, hogy glikoprotein. Korábban, egy hatásosnak ígérkező biokémiai tumor marker, az ún. lipidhez kötött sziálsav meghatározás (12, 13) elvi és módszertani alapjainak tisztázása során más szerzőkkel egybehangzóan megállapítottuk, hogy a próba nem a vérszérum lipidekhez kötött sziálsav tartalmát (gangliozidok, cerebrozidok), hanem a mintaelőkészítés sajátos körülményei között többé-kevésbé fajlagosan elválasztott, savanyú karakterű, igen jól oldódó és magas cukor (és sziálsav) tartalmú szérum mucoproteineket, elsősorban a vér savanyú alfa-1 glikoprotein (AGP) frakcióját mutatja ki (14, 15).
Az AGP (orosomucoid) a humán szérum egyik jellegzetes, 40 kD móltömegű, igen savanyú (pI=2-3), rendkívül jól oldódó és feltűnően magas (45%) szénhidrát tartalmú, akut fázis fehérje jellegű, szialoglikoprotein frakciója. Az AGP szénhidrát tartalma a lineáris peptid lánc 5 kitüntetett helyén (15-, 38-, 54-, 75-, 85) asparaginhoz N-glikozidos kötéssel kapcsolódó un. glikán egységekből áll. Az AGP-ben az oligoszacharid láncok elágazó (antennáris) szerkezetének megfelelő arányban a biopolimer össztömegére vonatkoztatva 8-10% galaktóz, 6-8 mannóz, kb. 1,0% fukóz, 12-16% hexózamin (N-acetil glukózamin) és 12-16% sziálsav (N-acetil neuraminsav) található. Az AGP molekulárisan heterogén (polimorf) és változatos oligoszacharid szerkezetéből adódóan különböző elválasztástechnikai módszerekkel (ioncserélő kromatográfia, lektin-immunoaffinitási elektroforézis, izoelektromos fókuszálás) molekula variánsokra választható szét. A Schmid és mtsai (21,22) által részletesen tanulmányozott és több mint 50 éve ismert AGP fiziko-kémiai tulajdonságairól, élettani szerepéről és biológiai aktivitásáról Kremer és mtsai (23) adtak 1988-ig igen részletes irodalmi áttekintést. A számos pathofiziológiai és kísérleti megfigyelésből általában megállapítható, hogy a humán szérum AGP tartalma egészséges egyéneknél 30-80 mg/dl határok között, diurnálisan is változó érték. A szérum AGP szintje nőknél, újszülötteknél, terhességben, fogamzásgátlók szedése és ösztrogén terápia esetén, valamint krónikus májbetegségekben az átlagosnál alacsonyabb. A szérum AGP tartalma, infekciók, gyulladások és traumák (égések, infarktus, sebészeti beavatkozások, általában akut fázis szindrómák) esetén, mint aspecifikus marker időlegesen és változatos mértékben megemelkedhet. A daganatos megbetegedések széles skálájában tartósan, a normálisnál jelentősen magasabb koncentrációk (150-400 mg/dl) mérhetők. A szérum AGP tartalmát, különösen más diagnosztikai próbákkal együtt a malignus folyamatok felismerésében, progressziójában és a terápia hatásosságának követésében, általában jól használható markernek tekintik (23-28). A szérum AGP túlnyomó része más akut fázis fehérjékhez hasonlóan elsősorban a májban szintetizálódik, de limfocita és epiteliális eredete is kimutatható. Akut fázis szindrómákban, és gyulladásokban a szérum AGP szint emelkedését elsődlegesen a májsejtek fehérjeszintézisének szintjén a makrofágokban keletkező citokinek (TNFa, IL-1, IL-6) transzkripciót stimuláló-mediáló hatásával magyarázzák. Miután a máj immun stimulációja és fokozott enzimindukciója a daganatos megbetegedéseknél egyértelműen nem igazolható, feltételezik, hogy a keringés esetenként igen magas, a normálistól szerkezetileg is eltérő AGP tartalma jelentős részben a degradálódó daganatsejtekből, ill. membránjaikból származik (29-34). Számos megfigyelés ellenére az AGP élettani szerepe sok tekintetben még ma is tisztázatlan. Megállapították, hogy mint elsődleges mitotikus inhibitor gátolja a sejtszaporodást és növekedést, valamint a limfociták rozetta képzését és mutagének, ill. allogének által indukált blasztogenezisét (36-38). Az immunválaszt módosító kettős (szuppressziv és induktív) hatása az AGP egyik sajátos biológiai aktivitását mutatja (35). A fentieken túlmenően feltételezhető, hogy mint igen savanyú hordozó (karrier) fehérje fontos szerepet tölt be a szteroidok, különböző kationos élettani vegyületek (pl. biogén aminok) és terápiás hatóanyagok kötésében, transzportjában, tárolásában (máj) és eliminációjában (23).
A szérum AGP szint meghatározására több konvencionális (immun diffuziós, -precipitációs, -turbiditási, LIA, RIA, ELISA,) módszer ismeretes (lásd a 3. Táblázatban). Korábban nagyhatékonyságú ioncserélő folyadékkromatográfiás (HPLC) mikroanalitikai módszert dolgoztunk ki és vezettünk be a szérum AGP tartalmának érzékeny és fajlagos mennyiségi mérésére (33). A normális (és kóros) értékhatárok vizsgálata során az irodalmi és saját adataink összevetése azt mutatta, hogy az egyes szerzők által alkalmazott módszerek, - többnyire fajlagosságuk mértékétől függően, - eltérő adatokat eredményezhetnek, míg a specifikus és pontosabb eljárások költségesek, vagy a rutinvizsgálatokra túl bonyolultak. Miután az általunk kidolgozott mikroanalitikai módszer (rövid leírása az Anyagok és Módszereknél) eszköz igénye (HPLC) miatt a kisebb és középszintű laboratóriumokban nem kivitelezhető, az alábbiakban részletezett módon gyors, egyszerű kolorimetriás eljárást fejlesztettünk ki és validáltunk az emberi vérszérum AGP tartalmának mennyiségi meghatározására.

Vizsgálatainkat klinikailag és laboratóriumi adatokkal igazolt egészséges egyének (közel azonos arányban mindkét nembeli, 19-57 év közötti), valamint az Országos Onkológiai Intézetben kezelt daganatos (elsősorban nőgyógyászati) betegek vérszérumából végeztük. A reggel, éhgyomorra, könyökvénából alvadásgátló nélkül vett, spontán megalvadt vérmintákból az alakos elemeket centrifugálással (5000 ford/perc, 0oC, 10 perc) választottuk el és a szérumokat a további felhasználásig -20oC-on tároltuk. A nagyobb mennyiségű mintát igénylő módszertani (validálási) vizsgálatok céljára gyűjtött (pooled) normál, ill. kóros szérum mintákat alkalmaztunk.
Vizsgálatainkban kétszer (üveg) desztillált vizet és az alábbiakban felsorolt analitikai tisztaságú (Reanal Finomvegyszergyár, Budapest) oldószereket és reagenseket alkalmaztuk: metil alkohol, kloroform, tetrahidrofurán, trietil amin, n-butil amin, jégecet (=100%), sósav (37%), kénsav (96%), foszforsav (2%), trifluoroecetsav, difenil amin, orcin, fenol, foszfor volfrámsav (P2O5.24WO3.nH2O), CaCl2.6H2O, Cd-acetát.2H2O, CdCl2.2H2O, CdSO4.8H2O, CoCl2..6H2O, CuSO4.5H2O, FeCl3.6H2O, HgCl2, MgCl2.6H2O, MnCl2.6H2O, NiCl2.6H2O, Zn-acetát.2H2O, ZnCl2 (vizm.), ZnSO4.7H2O. A fémion sók 1M vizes oldatát használtuk. A fehérjék teljes kicsapása 2 térf. mintához 1 térf. 2N sósavban oldott 5%-os foszfor volfrámsavval történt. A referencia szénhidrát oldatokat (1,0 mg/ml) min. 98%-os tisztaságú mannóz, galaktóz, fukóz, glukóz, sziálsav (N-acetil neuraminsav), glukózamin, galaktózamin (Sigma Co, St Louis Md) termékekből készítettük. Valamennyi kalibrálási és azonosítási eljárásban (HPLC, spektrofotometria, kolorimetria, elektroforézis) az általunk előállított és tisztított alfa-1-savanyú glikoproteint, valamint a kereskedelmi terméket (Acid alpha-1-glucoprotein, human, purified from Cohn-fraction VI, purity: 99%, Sigma Co, St Louis Md) használtuk.
A kolorimetriás mérések, az optikai elnyelési spektrumok felvétele, valamint a különböző minták fehérje (AGP) tartalmának meghatározása HITACHI U-2000 típusú precíziós spektrofotométerrel történt. A mintaelőkészítés, hidrolízis, származékkészítés, színreakció folyamataiban Labor MIM Vibroterm rázatott (jéggel hűtött) vízfürdőt és MTA KUTESZ 656 tip. Block-Therm, ill. 615 tip. kémcső termosztátokat alkalmaztunk.
Az AGP-fémion csapadékok szénhidrát tartalmának kolorimetriás mérésére kisebb módosításokkal a következő módszereket próbáltuk ki (40, 41):
Fenol-kénsavas reakció: 0,5 ml térfogatú mintához (50-100 µg cukor, v. 100-200 µg AGP) 3,0 ml fenol-kénsav reagenst adunk és összekeverés után 30 percig szoba hőmérsékleten tartjuk. A kialakult színintenzitást 490 nm-en reagens (vak) minta ellen fotometriával mérjük. Fenol-kénsav reagens: felhasználás előtt 1 térf. fenol 5%-os vizes oldatához óvatosan 5 térf. tömény kénsavat adunk, majd szobahőmérsékletre hűtjük. Orcin-kénsavas reakció: 0,5 ml térfogatú mintához 4,5 ml orcin reagenst adunk és összekeverés után kémcső termosztátban 20 percig 100oC-on hevítjük. A kialakult színintenzitást 430 nm-en reagens (vak) minta ellen fotometriával mérjük. Orcin-kénsav reagens: 1,4 g orcint oldunk 100 ml 60%-os kénsavban.
A difenil amin (DFA) színreakciót (40) a kísérleti eredményeink alapján módosítottuk, és a szérum AGP tartalom kolorimetriás meghatározására a következőképpen alkalmaztuk:. 400 µl humán szérumot csiszolatos üvegdugós (NS 14/23) centrifuga csőben (100x14 mm) 400 µl jéghideg desztillált vízzel hígítunk, majd 6,0 ml kloroform-metanol (2:1 v/v) oldószerelegyet adunk hozzá és 30 percig jeges vízfürdőben rázatjuk. Ezután 1,0 ml jéghideg desztillált vizet mérünk hozzá és a rázatást további 15 percig folytatjuk. A mintákat ezután 10 percig (0-4oC, 5000 ford/perc) centrifugáljuk. A vizes-metanolos felülúszó fázisból 500 µl-t csiszolatos dugójú centrifugacsőbe átemelünk és 100 µl 1 M ZnSO4 oldatot adunk hozzá. ™sszekeverés után (Vortex) 10 percig hűtőszekrényben (0-4oC) állni hagyjuk, majd a keletkező csapadékot lecentrifugáljuk (5000 ford/perc, 0-4oC, 10 perc). A felülúszó fázist és a cső falán lévő folyadékot lehetőség szerint (leszívással) teljesen eltávolítjuk. A csapadékot 1,0 ml jégecetben feloldjuk és 2,0 ml difenil amin reagenst mérünk hozzá. Erélyes felkeverés (Vortex) után a mintákat 1 órán át kémcső termosztátban 100oC-on tartjuk. A csövek gyors lehűtése után a kialakult színintenzitást 630 nm-en, reagens (vakpróba) ellen 10 mm-es üvegküvettában fotométeren mérjük. A szín intenzitása szobahőmérsékleten 3 órán belül nem változik. Difenil amin reagens: enyhe melegítés (40-50 C) közben 3,0 g difenil amint oldottunk 100 ml jégecet és 60 ml tömény (37%) sósav elegyében. A reagenst sötét üvegben, vagy adagoló pipettában (Oxford) tároltuk és kb. 2 hetente frissen készítettük.
A mérést galaktózt és mannózt (Gal 42 mg és Man 58 mg 100 ml desztillált vízben) tartalmazó, vagy 6,50 mg/ml AGP tartalmú törzsoldattal kalibráljuk. A Gal+Man törzsoldatból 5, 10, 20, 30, 40 és 50 µl-t üvegdugós centrifugacsövekbe mérünk, majd 1,0 ml jégecet és 2,0 ml difenil amin reagens hozzáadása után a színreakciót az előzőekben leírt módon folytatjuk. A szérum AGP tartalma a standard sorral meghatározott lineáris összefüggés alapján számítható. Sorozat vizsgálatoknál 20 µl Gal+Man standard, valamint a minták optikai elnyelését mérve (Astandard és Aminta) a szérum AGP tartalma a következő egyenlettel számítható:
AGP (mg/dl Szérum) = 198,4 x Aminta/Astandard
Az egyenlet szorzótényezője a közel monokromatikus optikai mérésre (spektrofotométer, 630 nm) vonatkozik. A mérés megfelelő, ha a 20 µl-es Gal+Man standardra kapott abszorbancia értéke A630=0,384±0.015. Egyszerűbb fotométerek, vagy koloriméterek (színszűrők) használata esetén a szorzótényezőt ellenőrizni, ill. módosítani kell.
A különböző minták fehérje (AGP) tartalmát az alábbiakban röviden ismertetett folyadékkromatográfiás (HPLC) módszerünkkel határoztuk meg (33). Vizsgálatainkban két P-500 pumpából, UV-1 monitorból (280 nm) és LCC-500 programozó-integráló egységből álló HPLC (Pharmacia, Uppsala, Svédország) folyadékkromatográfot alkalmaztunk. A minta előkészítése, a humán szérum oldószeres extrakciója az előzőekben részletezett módon történt. A szérum vizes-metanolos extraktumát (500 µl) közvetlenül a bis tris-propán pufferrel (50 mmol, pH 7.5, A-oldat) egyensúlyba hozott analitikai ioncserélő oszlopra (Fractogel EMD TMAE-650, 5x0,5 cm, 60-90 fim, Merck, Darmstadt) injektáltuk. A minták fehérje tartalmát kombinált NaCl-pH gradiens elucióval választottuk el, ahol a B oldat (50 mmol bis tris-propán, 350 mmol NaCl, pH 9,5) aránya 20 ml térfogat átáramlása során lineárisan 0-ról 100%-ra változott. A mérés kalibrálása az arbitrális csúcsmagasságok (AUSF 0,1) alapján történt.
Az AGP minták szénhidrát tartalmát folyadék~kromatográfiás (RP HPLC) módszerekkel analizáltuk. A sziálsav tartalmat Hara és munkatársainak eljárásával (52) határoztuk meg. A monoszacharidokat és hexózaminokat p-aminobenzoesav etilészter származékok formájában mértük (39).
Eredményeinket a lineáris korreláció számítás és a kétváltozós Student-t próba alapján értékeltük. Az egészséges (normál) szérum AGP értékek eloszlását (aszimmetria faktor) 20-120 mg/dl értékhatárok között 10 mg/dl intervallumokban grafikus módszerrel vizsgáltuk. A szérum AGP tartalom meghatározásának specificitását a normális felső határérték (cut off) alapján a következő összefüggés alapján számítottuk: specificitás = 100x(cut-off alatti értékek/összes érték)
Korábban nem elegyedő fázisú, oldószeres ex~trakción alapuló, egyszerű és gyors minta előkészítési eljárást dolgoztunk ki a humán szérum AGP tartalmának preparativ elválasztására, tisztítására és mikroanalitikai mérésére (33). A módszer előnye, hogy a szérum fehérjék túlnyomó részének (albumin) eltávolításával kíméletes körülményeket biztosít az AGP kinyerésére, és a vizes-metanolos fázis (extraktum) a preparatív, vagy analitikai ioncserélő folyadék kromatográfia céljaira közvetlenül felhasználható. Kimutattuk, hogy az extrakció optimált körülményei között (térfogat arányok, hőmérséklet, idő) a szérum AGP tartalmának több mint 96%-a változatlan összetétellel, bomlás nélkül a vizes-metanolos fázisba kerül és a vizes-metanolos extraktum 1,0 ml-e 130 µl szérummal egyenértékű. Az extraktum alkalmazása a kolorimetriás eljárás céljaira azonban szükségessé tette az AGP oldhatatlan fémion komplex formájában történő szelektív elválasztását a szérum egyéb (a vizes-metanolos fázisban oldódó) kromogén anyagaitól.
Bár az AGP ólom és cink sóját korábban kristályosan is előállították (22), a glikoproteinek fémion komplexeiről és oldékonyságáról, különösen összetett fehérje keverékekben viszonylag kevés adat ismeretes. A nehéz és átmeneti fémionok megfelelő kísérleti körülmények (ionerősség, pH, polaritás) között fehérjékkel oldhatatlan komplexeket képezhetnek és kicsapó reagensként alkalmazhatók. A szervetlen heteropolimer komplex fémsók között elsősorban a foszforvolframsavat (P205.24WO3.nH2O), mint teljes fehérje kicsapást eredményező reagenst a fehérjekémiában elterjedten alkalmazzák. Az ugyancsak általánosan használt triklór ecetsav, szulfoszalicilsav, perklórsav, ammónium szulfát, stb. a magas szénhidrát tartalmú, savanyú karakterű szialoglikoproteineket, így pl. az AGP-t tapasztalat szerint nem, vagy csak részlegesen precipitálják. Előzetes vizsgálataink azt mutatták, hogy a humán szérum vizes-metanolos extraktumának fehérje tartalma a referenciának tekinthető foszforvolframsavhoz viszonyítva a nehéz és átmeneti fémionok mellett (Cu2+, Co2+, Ni2+, Cd2+2+, Hg2+, Fe3+) részben alkáli földfémekkel (Ca2+, Mg2+, Ba2+) is kicsapható. A kolorimetriás módszer igényeit figyelembe véve a vizes-metanolos extraktum fehérje tartalmának kicsapására alkalmazható fémionok kiválasztását a következő szempontok határozták meg:
A fémion teljes fehérje kicsapást eredményezzen.
A reagens az AGP molekuláris (oligoszacharid) szerkezetét ne károsítsa.
A kolorimetriás kiértékelést ne zavarja.
A környezet szennyezés, toxicitás szempontjából a legkedvezőbb legyen.
A fémion csapadékok fehérje (AGP) tartalmának analízisévél (gélelektroforézis, HPLC) megállapítottuk, hogy a cink és kadmium sók 0,2 M végkoncentrációban teljes precipitációt adnak. A színes, nehéz és átmeneti fémionok (réz, kobalt, nikkel, vas) hasonló eredményre vezetnek, de meghamisítják a fotometriás kiértékelést. A Fe3+ és Hg2+ (és a foszforvolfrámsav) esetében kimutatható az AGP oligoszacharid szerkezetének károsodása (savas hidrolízis, oxidáció, oldható fragmentumok lehasadása), amely végső soron bizonytalanná teszi az AGP szénhidrát tartalmának alapján történő analitikai mérését. Tekintettel a kadmium ismert toxicitására, a kolorimetriás módszer céljaira végeredményben a cink sókat, választottuk. Tapasztalatunk szerint az anionok minősége (klorid, szulfát, acetát, nitrát) az oldhatatlan AGP-Zn komplex keletkezését nem befolyásolja.
A kolorimetriás mérés céljaira megfelelő színreakciót tiszta (kereskedelmi) AGP alkalmazásával és a módszereknél felsorolt reagensek összehasonlításával választottuk ki. Általában megállapítottuk, hogy mindhárom reagens esetében aránylag tág határokon belül (20-500 µg AGP) lineáris összefüggés van az optimális hullámhosszon mért optikai elnyelés (Amax) és az AGP mennyisége között. A színreakciók moláris abszorpciós koefficiensei (Amax mol x liter) fenol-kénsavra: A490 = 3,41x105, orcin-kénsavra: A430 = 13,83x105 értékeknek adódtak. A difenil amin esetében a minta vizes oldatával A630/víz = 6,27x105, míg jégecetben oldva A630/AcOH = 8,97x 105 értékeket kaptunk. Ez a megfigyelés azt mutatta, hogy a reakcióelegy savkoncentrációjának csökkentése (a minta víztartalma) csökkenti a difenil aminnal kialakuló szín intenzitását. Ezért továbbiakban a kolorimetriás mérés céljaira a különböző mintákat (AGP-fémion csapadékok, monoszacharid standardok) jégecetben oldottuk. A színreakciók reprodukálhatóságát, érzékenységét, a reagensek viszkozitását és korroziv tulajdonságait figyelembe véve a kolorimetriás mérésre végső soron a difenil amin színreakciót találtuk legalkalmasabbnak.
A kolorimetriás eljárásunkban alkalmazott difenil amin színreakciót és az AGP szénhidrát összetevőinek viselkedését részletesebben tanulmányoztuk. A színreakció mechanizmusában az erősen savas közeg megfelelő hőmérsékleten először megbontja az oligoszacharidok szerkezetét, majd a glukozidos kötésekből felszabaduló monoszacharidok a difenil aminnal reakcióba lépnek. Ismeretes, hogy a különböző egyszerű és összetett szacharidok a kolorimetriás színreakciókban jelentősen eltérő fajlagos színintenzitást eredményeznek (40,41). Az AGP szénhidrát összetevőinek ismeretében összehasonlítottuk az egyes cukor komponensek fajlagos színintenzitását. Az 1. ábra mutatja, hogy az AGP oligoszacharid szerkezetében számításba vehető monoszacharidok közül a galaktóz és mannóz nagyságrenddel nagyobb színintenzitást eredményez, mint a többi összetevő (fukóz, hexozaminok, sziálsav). Az AGP glukózt nem tartalmaz. Tekintetbe véve az AGP oligoszacharid tartalmának összetételét (l. később) megállapítható, hogy a difenil amin színreakciót gyakorlatilag csak a biopolimer galaktóz és mannóz tartalma hozza létre. További vizsgálataink alátámasztották, hogy az AGP mennyiségi meghatározása megfelelő arányú galaktóz-mannóz keverékkel kalibrálható.
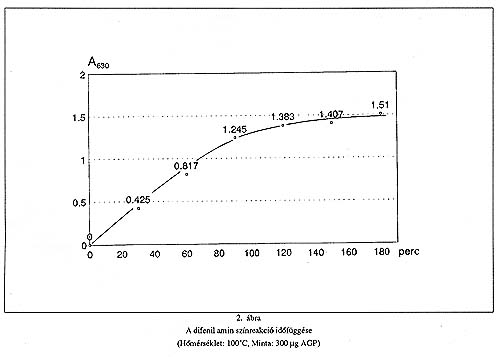
A színreakció optimális paramétereinek meghatározása során megállapítottuk, hogy a reagens jégecet és sósav tartalma miatt a hevítés hőfoka 100oC-nál magasabb nem lehet. A 2. ábra a hevítés időtartamának vizsgálatát mutatja. Látható, hogy a színintenzitás 120 percig folyamatosan növekszik, a reakcióidő további növelése számottevő előnyt nem jelent. A módszer időigényének csökkentése érdekében a hevítés időtartamát célszerűen 60 percnek választottuk. A 3. ábra a difenil amin színreakció stabilitását szemlélteti. Megállapítható, hogy a 60 perc alatt 100oC-on kialakult színintenzitás szobahőmérsékleten további 3 órán keresztül számottevő változást nem mutat.
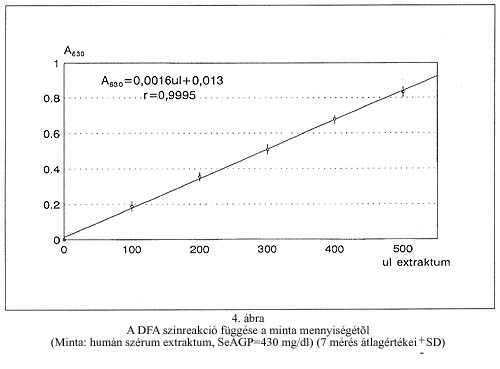
A kolorimetriás mérés linearitásának, a mérendő anyagmennyiség (AGP) és a keletkező színintenzitás összefüggésének vizsgálata céljából magas (430 mg/dl) AGP tartalmú, gyűjtött humán szérumból a minta előkészítés lépéseiben leírt módon vizes-metanolos extraktumot készítettünk. Az extraktumból csökkenő térfogatú (500, 400, 300, 200 és 100 µl) részletekkel a további műveleteket (fémion kicsapás, színreakció) elvégezve úgy találtuk (4. ábra), hogy a humán szérumban mérhető AGP koncentráció tartományban az optikai elnyelés (A630) a bemért anyagmennyiséggel lineáris összefüggést ad. Gyakorlatilag tehát a kolorimetriás módszer ismert AGP tartalmú standard szérummal is kalibrálható.
A kolorimetriás mérés hitelesítésére (kalibrálására) referencia vegyületként elsősorban kereskedelmi AGP-t alkalmaztunk. A komplex makromolekula fiziko-kémiai tulajdonságainak vizsgálata azonban azt mutatta, hogy a pontos standard oldat elkészítésének elvi és gyakorlati akadályai lehetnek. A magas szénhidrát tartalmú, rendkívül hidrofil karakterű biopolimer változó mennyiségű, szerkezetileg kötött vizet tartalmazhat, amely a súly szerinti bemérést pontatlanná teheti. Bomlást és egyéb kontaminánsok jelenlétét feltételezve egyes esetekben a kereskedelmi termék hatóanyag tartalma is kérdésesnek bizonyult. A súly szerint bemért standard oldat AGP tartalmának ellenőrzése más, konvencionális analitikai módszerekkel (Folin-Lowry, UV-abszorpció, Kalckar-formula) elvi okokból is bizonytalan. Az AGP szénhidrát összetevői (elsősorban a sziálsav) a fehérje meghatározási módszereket eltérő és nehezen kompenzálható módon torzítják. Végső soron az analitikai módszerek korrekt kalibrálása csak a gondosan tisztított és szárított, minőségileg ellenőrzött AGP súlyszerinti bemérésével valósítható meg. Tekintetbe véve a fenti körülményeket, és a kereskedelmi AGP viszonylag magas árát a kolorimetriás módszer kalibrálására (validálására) az AGP szénhidrát összetevőinek, elsősorban galaktóz és mannóz tartalmának mérésén alapuló eljárást dolgoztunk ki. Előzőekben kimutattuk, hogy a difenil aminnal kialakuló színreakció szinte kizárólag az AGP galaktóz és mannóz tartalmának tulajdonítható, és a két monoszacharid fajlagos színintenzitása egymástól jelentősen különbözik. A galaktóz-mannóz eleggyel történő kalibrálás alátámasztására saját anyagunkban 14 egészséges egyén és 18 daganatos (uterus és ovarium cc) beteg szérumából kinyert és tisztított AGP mintában meghatároztuk a szénhidrát tartalmat és a cukor összetevők arányát. Az irodalmi adatokkal összhangban (21,23,50,53,54) úgy találtuk, hogy a szérumok egészséges, vagy daganatos eredetétől függetlenül az AGP oligoszacharid tartalma viszonylag állandó érték, átlagosan 45%. Az AGP monoszacharid összetevőinek HPLC analízise során nem találtunk szignifikáns különbséget az egészséges és daganatos minták galaktóz, mannóz és hexózamin (N-acetil glukózamin) tartalma között. Megállapítottuk, hogy a galaktóz (8,9±2,0%) és mannóz (6,6±1,5%) együttes mennyisége átlagosan az AGP 15,5%-át teszi ki és arányuk ennek megfelelően Gal/Man = 1,35. A hexózaminok közül az átlagosan 13,5±2,7% N-acetil glukózamin mellett N-acetil galaktózamin 1%-nál kevesebb volt. Magasabb (gyengén szignifikáns, p<0.05) sziálsav és fukóz tartalmat mértünk a daganatos betegeknél (15,6±3,7 % ill. 1,7±0,3%) az egészséges AGP minták (11,5±3,0% ill. 1,0±0,2% ) értékeihez képest.

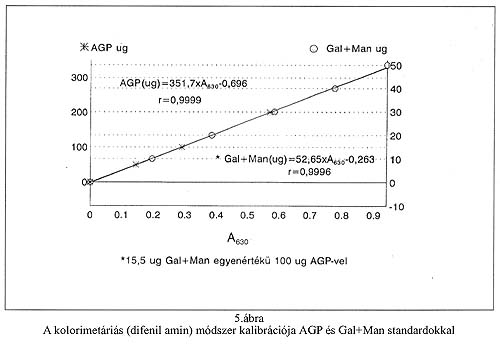
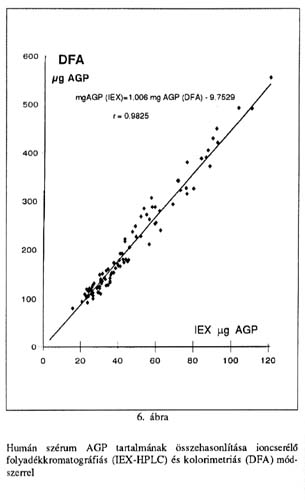
A kolorimetriás módszer kalibrálására vonatkozóan a fentiekből következik, hogy 15,5 µg galaktóz+mannóz keverék, amely arányaiban 57,5% galaktózt és 42,5% mannózt tartalmaz 100 µg AGP-vel egyenértékűnek tekinthető. A tiszta AGP-vel és a galaktóz-mannóz standard oldattal kapott mérési eredményeket (7-7 vizsgálat sorozat átlagértékei ±SD) az 1. és 2. Táblázatok tüntetik fel. Az 5. ábrán látható, hogy az AGP-vel és a galaktóz-mannóz standard oldattal felvett kalibrációs mérési pontok tág értéktartományban (20-500 µg AGP) lényegében azonos, lineáris összefüggéseket eredményeznek. A kolorimetriás módszer érzékenysége (kimutathatósági-mérési alsó határ) 15 µg AGP (2 µg galaktóz+mannóz), amely megfelel <126>20 mg/dl szérum koncentrációnak. A kolorimetriás módszer kalibrálása galaktóz-mannóz standard oldat, vagy gyűjtött humán szérum ismételt (day to day) bemérésével átlagosan 5,4%-os pontossággal (C.V.) reprodukálható. Az 1. és 2. Táblázat összevetéséből azonban kitűnik, hogy a mérés reprodukálhatósága a galaktóz-mannóz standard oldattal jobb (átl. 2,95% C.V.), mint AGP alkalmazásával (átl. 9,53% C.V.). A humán szérumok individuális tulajdonságaiból (kromogén kontaminánsok, gyógyszerek) adódó esetleges eltérések kimutatására egészséges egyénektől és daganatos betegektől származó, összesen 100 egyedi szérum minta AGP tartalmát a difenil aminos kolorimetriás és az ioncserélő folyadékkromatográfiás módszerrel párhuzamosan meghatároztuk. A 6. Ábra mutatja, hogy a két eljárás jó korrelációval (r=0,9857) közel azonos eredményt ad.

Vizsgálatainkból végső soron arra következtethetünk, hogy a minta előkészítés lépésein (extrakció, fémionos kicsapás) és a difenil amin színreakción alapuló kolorimetriás mérési módszer a viszonyítási alapnak tekinthető ioncserélő folyadékkromatográfiás eljárással egyenértékű és az analitikai mérés hibahatárán belül azonos eredményt ad. Az AGP oligoszacharid szerkezetére, ill. a difenil amin színreakció körülményeire vonatkozó HPLC és spektrofotometriai méréseink alátámasztották, hogy az 57,5% galaktózt és 42,5% mannózt tartalmazó standard oldat a kolorimetriás módszer kalibrálására alkalmas, és az AGP-nél jobban használható.
A kolorimetriás módszer rutin laboratóriumi alkalmazhatóságát normál humán populációban és daganatos beteganyagon vizsgáltuk. Korábban beszámoltunk a szérum AGP meghatározás diagnosztikai értékéről a különböző gasztroenterológiai tumorok felismerésében (28). Nagyobb, elsősorban nőgyógyászati beteganyagra vonatkozó megfigyeléseink feldolgozása folyamatban van. Munkánk során 1995 és 1999 között két egymástól független vizsgálat sorozatban (n1=40 és n2=35) összesen 75 egészséges egyén szérum AGP tartalmának meghatározása átlagosan 59,9±16,2 mg/dl értéket eredményezett. Az adatok az átlagnál magasabb értékek felé eltolódó, enyhén aszimmetrikus eloszlást mutattak (aszimmetria faktor: 1,20). A normális felső határérték (cut-off) a specificitás meghatározásánál ajánlott átlagérték ±2SD számítást figyelembe véve 92,3 mg/dl-nek adódott. A szérum AGP meghatározás specificitását a cut-off érték függvényében vizsgálva adatainkra vonatkozóan megállapítottuk, hogy 76,1 (±SD), ill. 92,3 (±2SD) mg/dl cut-off értékeknél a próba specificitása a normál populációra 85%, ill. 98,7%. A gyakorlatban 80 mg/dl cut-off értéket választva a szérum AGP tartalom meghatározásának specificitása egészséges egyénekre 89%. A normális és kóros értékekre és a különböző mérési módszerekre vonatkozó 1988-ig fellelhető adatokat igen részletesen Kremer és mtsai foglalták össze (23). A 3. Táblázatban az elmúlt évtized hivatkozásaiból néhány további, módszertanilag elfogadható és~ mikroanalitikai szempontból is értékelhető eredményt tüntettünk fel. A felsorolásból kitűnik, hogy a mérések túlnyomó része immunológiai (diffúzió, elektroforézis, nefelometria/turbidimetria) módszerekkel történt. Eredményeinket a 3. Táblázat adataival összevetve megállapítható; hogy az egészséges egyéneknél az általunk kidolgozott ioncserélő folyadék~kromatográfiás és/vagy kolorimetriás módszerrel meghatározott átlagos szérum AGP tartalom és határértékei az irodalmi adatokkal jól egyeznek.
Az utóbbi évtizedek klinikai és kísérletes kutatásai a figyelmet fokozottan a keringés és a sejtmembránok szerkezetileg és funkcionálisan ubiquiter összetevőire, a szénhidrát tartalmú komplex fehérjékre irányították. A számos élettanilag fontos és biológiailag aktív szialoglikoprotein között az eddig ismert és többé kevésbé specifikus celluláris (tumor associated) és szerológiai markereknek (akut fázis fehérjék, antigének, metabolitok, stb.) a daganatos megbetegedések felismerésében és követésében elméleti és gyakorlati szempontból jelentős szerepe van. Figyelembe véve az ide vonatkozó irodalmi hivatkozások tekintélyes számát, a változatos molekula tömegű és szénhidrát tartalmú szialoglikoproteinek sorában a humán szérum alfa-1-savanyú glikoprotein (AGP) feltűnően magas és jellegzetesen strukturált oligoszacharid tartalma, rendkívül savanyú karaktere és oldékonysága, nem utolsó sorban sajátos élettani szerepe miatt láthatóan kitüntetett helyzetben van. Irodalmi adatok és saját megfigyeléseink is alátámasztják, hogy a humán szérum AGP tartalmának vizsgálata a daganatos megbetegedésekben biokémiai tumor markerként alkalmazható. A mennyiségi változáson túlmenően egyre több megfigyelés alátámasztja, hogy különböző (pl. daganatos) megbetegedések a szialoglikoproteinek glikán szerkezetében és szénhidrát összetételében jellegzetes - részleteiben még nem teljesen tisztázott - változásokat (pl. fokozott sziálsav és fakóz beépülést) indukálnak (16-18,42,45,46,51). A humán szérum AGP monoszacharid összetevőinek HPLC analízise, elsősorban galaktóz és mannóz tartalmának meghatározása és a szénhidrát szerkezetet felépítő cukrok tanulmányozása a difenil amin színreakcióban lehetővé tette a megfelelő arányú galaktóz-mannóz (Gal/Man=1,35) standard keverék alkalmazását a kolorimetriás módszer kalibrálására. Az AGP-vel és a galaktóz-mannóz standarddal elvégzett kalibrálás összehasonlítása igazolta, hogy a két eljárás egyenértékű, és a kalibrálás a galaktóz-mannóz standarddal jobban reprodukálható.
Eredményeinket a következőkben foglalhatjuk össze:
Nem elegyedő fázisú oldószeres extrakción és oldhatatlan fémion (Zn) komplex képzésen alapuló minta előkészítési módszert dolgoztunk ki és alkalmaztunk a humán szérum AGP tartalmának elválasztására és az egyéb kromogén kontaminánsok eltávolítására.
Az AGP szénhidrát tartalmának alapján történő meghatározása céljából összehasonlítottuk a mono- és oligoszacharidok kimutatására általánosan használt fenol-kénsav, orcin-kénsav és difenil amin reagenseket. A difenil amin színreakció kísérleti körülményeinek optimálásával kolorimetriás módszert dolgoztunk ki a szérum AGP tartalmának mennyiségi mérésére.
A színreakció mechanizmusának és az AGP monoszacharid összetevőinek szisztematikus vizsgálatával megállapítottuk, hogy a difenil amin reagenssel lényegében csak a galaktóz és mannóz adnak 630 nm-en jól mérhető, de fajlagosan különböző színintenzitást. Miután a többi cukor komponens hatása elhanyagolható, a difenil amin reagenssel kialakuló színintenzitást az AGP oligoszacharid szerkezetében a galaktóz és mannóz moláris (súly) arányainak megfelelő abszorpciós inkrementumok összege képezi.
Az AGP szénhidrát tartalmának vizsgálata céljából egészséges egyének és daganatos betegek szérumából izolált AGP mintákban HPLC módszerekkel meghatároztuk a monoszacharid összetevők minőségét és arányát. Mások megfigyeléseivel összhangban megállapítottuk, hogy az AGP minták egészséges, vagy kóros eredetétől függetlenül az oligoszacharidok antennáris szerkezetében a galaktóz és mannóz aránya viszonylag állandó (Gal/Man=1,35) és a galaktóz és mannóz együttes mennyisége jó közelítéssel a biopolimer össztömegének 15,5%-át képezi.
A fenti megfigyelések alapján megfelelő súlyarányban galaktózt (57,5%) és mannózt (42,5%) tartalmazó standard oldatot alkalmaztunk a szérum AGP tartalom kolorimetriás meghatározásának kalibrálására, amely a referencia vegyület (tiszta AGP) használatával egyenértékűnek bizonyult.
Egyedi humán szérum minták AGP tartalmának párhuzamos meghatározása a kolorimetriás módszerrel és az ioncserélő folyadékkromatográfiás mikroanalitikai eljárással gyakorlatilag azonos eredményeket adott.
Egészséges egyének szérum AGP tartalmának meghatározása a kolorimetriás (és/vagy ioncserélő kromatográfiás) módszerrel az irodalmi adatokkal egyező, átlagosan 59,9±16,2 mg/dl értéket adott. A mérési adatok statisztikai analízise alapján a szérum AGP meghatározás specificitása 80 mg/dl cut-off érték alkalmazása esetén egészséges populációra 89%.
Az Országos Laboratóriumi Intézet által jóváhagyott módszert a Reanal Finomvegyszergyár Rt - AGP Szérum alfa-glikoprotein Diagnosztikai Reagens Készlet - elnevezéssel hozza forgalomba.
Vizsgálatainkat az OMFB 94-97-65-0709 sz. és OTKA 021201 sz. kutatási keretei támogatták. Köszönetet mondunk Némethné Ary Zsuzsa, Susztrik Beatrix, Kovácsné Nikl Erika, Akacs Beáta és Kovács Margit szakszerű közreműködéséért.
Newlands ES: Clinical applications of tumor markers Med Lab Sci 1987; 44: 361-370
Chu TM: Biological markers for human cancer Curr Top Pathol 1987; 77: 19-45
Torosian MH: The clinical usefulness and limitations of tumor markers Surg Gynecol Obstet 1988; 166: 567-579
Klapdor R (ed) Recent Results in Tumor Diagnosis and Therapy, 5th Symp on Tumor Markers, Hamburg, W Zuckschwerdt Verlag, München, Bern, Wien, San Francisco. 1989
Fateh-Moghadam A Stieber P: Tumormarker und ihr sinnvoller Einsatz Jürgen Hartmann Verlag GmbH, Marloffstein-Rathsberg. 1993
Wu JT and Nakamura RM: Human Circulating Tumor Markers, Current Concepts and Clinical Applications ASCP Press, Chicago. 1997
Schumann B: Keringő tumor markerek klinikai alkalmazásai BYK Diagnosztika, Budapest, 1998
Berger EG, Buddecke E, Kamerling JP, et al.: Structure, biosynthesis and functions of glycoprotein glycans Experientia 1982; 38: 1129-1258
Hounsell EF: Physicochemical analysis of oligosaccharide determinants of glycoproteins Adv Carbohydr Chem Biochem 1994; 50: 311-350
Bonfichi R, Sottani C, Colombo L, et al.: Preliminary investigation of glycosylated proteins by capillary electrophoresis and capillary electrophoresis/mass spectrometry using electrospray ionization and by matrix-assisted laser desorption ionization/time of flight mass spectrometry J Mass Spectrom Rapid Comm Mass Spectrom 1995; 9: S95-S105
Oliver RWA Green BN Harvey DJ: The use of electronspray ionization MS to determine the structure of glycans in intact glycoproteins Biochem Mass Spectr 1996; 24: 917-927
Katopodis N Stock CC: Improved method to determine lipid bound sialic acid in plasma or serum. Res Commun Chem Pathol Pharmacol 1980; 30: 171-180
Katopodis N, Hirschaut Y, Geller NL, Stock CC: Lipid-associated sialic acid test for the detection of human cancer Cancer Res 1982; 42: 5270-5273
Voigtman R, Pokorny J, Meinshausen A: Evaluation and limitations of the lipid-associated sialic acid test for the detection of human cancer Cancer 1989; 64: 2279-2283
15. Végh Zs, Kremmer T, Boldizsár M et al.: A re-evaluation of the lipid-bound sialic acid determination Clin Chim Acta 1991; 203: 259-268
Van Dijk W. Havenaar EC Brinkman-Van der Linden ECM: <F128M>a<F255D>1-Acid glycoprotein (orosomucoid): pathophysiological changes in glycosylation in relation to its function Glycoconjugate J 1995; 12: 227-233
Dall'Olio F: Protein glycosylation in cancer biology: an overview J Clin Pathol Mol Pathol 1996; 49: M126-M135
Hounsell EF, Young M. Davies MJ: Glycoprotein changes in tumours: a renaissance in clinical applications Clin Sci 1997; 93: 287-293
Taylor CM: Glycopeptides and glycoproteins: focus on the glycosidic linkage Tetrahedron 1998; 54: 11317-11362
Drickamer K, Taylor M: Evolving views of protein glycosylation TIBS 1998; 23: 321-324
Schmid K: a1-Acid glycoprotein. In: Putnam PW (ed.): The plasma proteins. pp 183-228 Academic Press, New York, 1975
Schmid K: Human plasma alpha-1-acid glyco~protein-Biochemical properties, the amino acid sequence and the structure of the carbohydrate moiety, variants and polymorphism. In: Baumann P Müller WE Eap CB Tillement JP (eds) Alphal acid glycoprotein Genetics, Biochemistry, Physiological Functions and Pharmacology pp 7-22 Alain R Liss, Inc New York, 1989
Kremer JMH, Wilting J, Janssen LMH: Drug binding to human alpha-1-acid glycoprotein in health and disease Pharmacol Rev 1988; 40: 1-47
Ganz PA Mo PY Wong HJ et al.: Evaluation of three biochemical markers for serially monitoring the therapy of small-cell lung cancer J Clin Oncol 1987; 5: 472-479
Turner GA, Skillen AW, Buamah P, et al.: Relation between raised concentrations of fucose, sialic acid and acute phase proteins in serum from patients with cancer: choosing suitable serum glycoprotein markers J Clin Pathol 1985; 38: 588-592
Patel PS, Adhvaryu SB, Balar DB, et al.: Clinical application of serum level of sialic acid, fucose and seromucoid fraction as tumor markers in human leukemias Anticancer Res 1994; 14: 747-752
Charet JC, Watine J, Lepretre A, et al. Orosomucoid:prealbumin ratio in the monitoring of lung cancer Clin Biochem 1996; 29: 287-290
Szilvás Á, Székely Gy, Kremmer T, et al.: A szérum savanyú alfa-1 glikoprotein (AGP) meghatározása gastrointestinális tumorokban Orvosi Hetilap 1999; 139: 2199-2202
Harvey HA, Lipton A, White D, et al.: Glycoproteins and human cancer. II. Correlation between circulating level and disease status Cancer 1981; 47: 324-327
Hakomori SI: Aberrant glycosylation in cancer cell membranes as focused on glycolipids: overview and perspectives Cancer Res 1985; 45: 2405-2414
Shamberger RJ: Evaluation of water soluble and lipid soluble sialic acid level as tumor markers Anticancer Res 1986; 6: 717-720
Riley M, Tautu C, Verazin G, et al.: Evaluation of sialic acid concentrations in serum for the diagnosis and staging of breast cancer Clin Chem 1990; 36: 161-162
Kremmer T, Boldizsár M, Kovács J, et al.: Determination and analysis of human serum alpha-1-acid glycoprotein by liquid chromatographic methods J Liquid Chromatogr 1995; 18: 1207-1218
Kremmer T, Oláh S, Boldizsár M, et al.: New trends in the analysis and diagnostic application of sialoglycoprotein markers J Tumor Marker Oncol 1999; 14: 52-53
Su SJ, Yang BC, Wang YS, et al.: Alpha-1-acid glycoprotein induced tumor necrosis factor-a secretion of human monocytes is enhanced by serum binding proteins and depends on protein tyrosine kinase activation Immunopharmacology 1999; 41: 21-29
Onda H: a1-acid glycoprotein and a1-antitrypsin as mitotic inhibitors in regenerating rat liver Gann 1977; 68: 301-306
Kamik I, Gerber J, Dobryszycka W: Microheterogeneity of a1-acid glycoprotein in the sera of patients with cancer or inflammatory states of the ovaries Arch Immun Ther Exp 1988; 36: 1-6
Curtin NJ, Newell DR, Harris AL: Modulation of dipyridamole action by a1-acid glycoprotein Biochem Pharmacol 1989; 36: 3281-3288
Kwon H, Kim J: Determination of monosaccharides in glycoproteins by reversed-phase high-performance liquid chromatography Anal Biochem 1993; 215: 243-252
Vejdélek ZJ, Kakác: Farbreaktionenin der spektrofotometrischen Analyse organischer Verbindungen Band I. Organische Farbreagenzien pp 24, 69, 403 VEB Gustav Fischer Verlag Jena, 1969
Saha SK, Brewer CF: Determination of the concentrations of oligosaccharides complex type carbohydrates, and glycoproteins using the phenol-sulfuric acid method Carbohydrate Res 1994; 254: 157-167
Hansen JES, Larsen VA, Bog-Hansen TC: The microheterogeneity of a1-acid glycoprotein in inflammatory disease, cancer of the lung and normal health Clin Chim Acta 1984; 138: 41-47
Blain PG, Mucklow JC, Rawlins MD, et al.: Determinants of plasma a1-acid glycoprotein (AAG) concentrations in health Brit J Clin Pharmacol 1985; 20: 500-502
Yap AKL, Fish RG, Keen CW: Acute phase glycoproteins in sera of patients with sarcomas receiving methotrexate infusion therapy Clin Biochem 1985; 18: 70-72
Mackiewicz A, Marcinkowska-Pieta R, Ballon S, et al.: Microheterogeneity of alphal-acid glycoprotein in the detection of intercurrent infection in systematic lupus erythematosus Arthritis and Rheumatism 1987; 30: 513-518
Fassbender K, Zimmerli W, Kissling R, et al.: Glycosylation of a1-acid glycoprotein in relation to duration of disease in acute and chronic infection and inflammation Clin Chim Acta 1991; 203: 315-328
Lindberg G, Rastam L, Gullberg B, et al.: Serum concentrations of total sialic acid and sialoglycoproteins in relation to coronary heart disease risk markers Atherosclerosis 1993; 103: 123-129
Crook MA, Tutt P, Simpson H, Pickup JC: Serum sialic acid and acute phase proteins in type 1 and type 2 diabetes mellitus Clin Chim Acta 1993; 219: 131-138
Woo J, Chan HS, Or KH, et al.: Effect of age and disease on two drog binding proteins: albumin and a-1-acid glycoprotein Clin Biochem 1994; 27: 289-292
Kishino S, Nomura A, Sugawara M, et al.: Purification method for a-1-acid glycoprotein with subsequent high-performance liquid chromatographic determination of monosaccharides in plasma of healthy subjects and patients with renal insufficiency J Chromatogr B 1995; 672: 199-205
Kishino S, Nomura A, Di ZS, et al.: Alpha-1-acid glycoprotein concentration and the protein binding of disopyramide in healthy subjects J Clin Pharmacol 1995; 35: 510-514
Hara S, Takemori Y, Yamaguchi M, et al.: Fluorometric high performance liquid chromatography of N-acetyl- and N-glycolyl neuraminic acids and its application to their microdetermination in human and animal sera, glycoproteins and glycolipids Anal Biochem 1987; 164: 138-145
Tamura K, Shibata Y, Matsuda Y, et al.: Isolation and characterization of an immunosuppressive acidic protein from ascitic fluid of cancer patients Cancer Res 1981; 41: 3244-3252
Eggert FM, Jones M: Measurement of neutral sugars in glycoproteins as dansyl derivatives by automated high-performance liquid chromatography J. Chromatogr 1985; 333: 123-131
* Levélcím: Országos Onkológiai
Intézet
H-1122 Ráth Gy. u. 8-11